МЕТРОЛОГІЯ ФАРМАЦЕВТИЧНОГО АНАЛІЗУ — вчення про математичне (зокрема статистичне) оброблення результатів і є частиною метрології хімічного аналізу. Особливістю фармацевтичного аналізу є те, що його результат залежить від типу об’єкту, який аналізується (субстанція чи певна лікарська форма), та належить, як правило, до серійної продукції, а допуски вмісту компонентів, які аналізуються, є досить вузькими. Метрологічні характеристики фармацевтичного аналізу — невизначеність (для даної довірчої вірогідності), збіг, відтворюваність, межа виявлення тощо й оцінюються для методики аналізу конкретного об’єкта.
Можна виділити декілька основних типових завдань М.ф.а.:
1. Предмет статистичного аналізу. Це статистичне оброблення результатів аналізу й отримання метрологічних характеристик. М.ф.а., як правило, залежить від декількох випадкових величин (зважування, розведення розчинів, вимірювання і т.п.), тому загальна невизначеність таких методик є функцією декількох випадкових змінних. Взаємозв’язок загальної невизначеності М.ф.а. може бути виявлений за допомогою різних підходів, серед яких найбільш простим і зручним є лінійна модель. Згідно з нею загальна невизначеність результату ФА (∆Аs) може бути обчислена за формулою:
 , (1)
, (1)
де ∆i — окрема складова невизначеності, виражена як однобічний відносний довірчий інтервал для рівня надійної ймовірності 95%, у відсотках.
Зазвичай повну невизначеність результатів аналізу ∆Аs можна розбити на складові, пов’язані з невизначеністю пробопідготовки (∆SP) і з невизначеністю кінцевої аналітичної операції (∆FAO):
 (2)
(2)
У разі непрямих методів аналізу в це рівняння слід додати ще невизначеність стандартного зразка (∆st). Але згідно з концепцією застосування стандартних зразків у фармацевтичному аналізі атестація їх проводиться таким чином, щоб невизначеність приписного значення була незначущою порівняно з граничною загальною невизначеністю методик аналізу, для яких цей стандартний зразок застосовується. Тому його можна не враховувати в рівнянні (2). З іншого боку, для непрямих методів аналізу загальну невизначеність результатів аналізу можна розбити на складові частини, пов’язані з випробуваним зразком (sample) і стандартним зразком (st):
 . (3)
. (3)
2. Метрологічне обґрунтування методики фармацевтичного аналізу. Методики фармацевтичного аналізу, як і всі інші аналітичні методики, повинні бути метрологічно обґрунтовані й метрологічно забезпечені. Метрологічна обґрунтованість конкретної аналітичної методики означає, що її невизначеність є прийнятною для вирішення поставлених завдань. Якщо це не виконується, то така невизначеність суттєво впливає на прийняття рішень про якість ЛП, що аналізується. Це означає, що в різних лабораторіях можуть бути отримані різні результати, які статистично не розрізняються, але приведуть до різних висновків про якість. Метрологічне обґрунтування конкретної методики контролю якості ЛП є одним з обов’язкових етапів проведення валідації аналітичних методик і випробувань. Метрологічна забезпеченість конкретної аналітичної методики в конкретній аналітичній лабораторії означає, що експлуатаційні характеристики аналітичного обладнання (терезів, спектрофотометрів, хроматографів тощо) і мірного посуду є достатніми для виконання аналізу за конкретною методикою конкретної АНД. Метрологічна забезпеченість є одним з етапів кваліфікації обладнання на стадії валідації аналітичних методик і випробувань. При контролі якості ЛП зазвичай спираються на фармакопейні вимоги до аналітичного обладнання й мірного посуду, які вважаються достатніми для виконання методики АНД, якщо в них немає інших вказівок. При проведенні метрологічного обґрунтування аналітичної методики і визначення її метрологічної забезпеченості спираються на прогноз загальної невизначеності методики аналізу (або їх складових частин). При цьому використовують граничні невизначеності експлуатаційних характеристик обладнання й мірного посуду згідно з ДФ або АНД.
Типовими методиками більшості фармакопейних методів є рідинна хроматографія і спектрофотометрія. Але схожі проблеми виникають для всіх інших кількісних методів фармацевтичного аналізу. Для спрощення пробопідготовка для обох методів зроблена однаковою. Згідно з АНД при проведенні методики кількісного визначення речовини Х в ЛП Z беруть наважку 10 мг ЛП Z, уміщують її в мірну колбу місткістю 10 мл, доводять розчинником до мітки (випробуваний розчин). Паралельно готують розчин порівняння: 50 мг фармакопейного стандартного зразка (ФСЗ) речовини Х уміщують у мірну колбу місткістю 25 мл. 1,0 мл отриманого розчину вміщують у мірну колбу місткістю 50 мл і доводять розчинником до мітки.
Рідинна хроматографія. Поперемінно хроматографують випробуваний розчин і розчин порівняння на рідинному хроматографі, отримуючи не менше ніж по 5 хроматограм кожного розчину. Розрахунок вмісту речовини Х в ЛП проводять методом стандарту. Згідно з тестом на придатність хроматографічної системи відносне стандартне відхилення площ піків (RSDmax), розраховане з 5 хроматограм стандартного розчину, має бути не більше 0,90%.
Спектрофотометрія. Проводять по 3 вимірювання оптичної густини випробуваного розчину і розчину порівняння з рандомізацією положення кювет. Усереднюють результати і розраховують вміст речовини Х в ЛП Z, виходячи з концентрації розчину порівняння. Чи є такі методики контролю речовини Х у ЛП Z метрологічно обґрунтованими, якщо допуски вмісту речовини Х у ЛП Z становлять 95,0–105,0% від номінального вмісту речовини Х? Для вирішення цього питання необхідно зробити прогноз загальної невизначеності методики аналізу та порівняти її з критерієм прийнятності. У свою чергу, прогноз загальної невизначеності методики аналізу поділяється на прогноз невизначеності пробопідготовки і прогноз невизначеності кінцевої аналітичної операції.
Таким чином, типова задача метрологічного обґрунтування методики фармацевтичного аналізу містить такі етапи:
2.1. Прогноз граничної невизначеності пробопідготовки (∆SP). При прогнозі граничної невизначеності пробопідготовки методик фармацевтичного аналізу доцільно спиратися на фармакопейні вимоги до невизначеності мірного посуду і терезів, приймаючи ці невизначеності за довірчі інтервали з вірогідністю 95%. Зокрема, гранична невизначеність різних операцій для вищенаведеного прикладу типової задачі становить :
ФСЗ (st): наважка 50 мг Х — 0,2 мг, або 100·0,2/50=0,40%, мірна колба місткістю 25 мл — 0,23%, піпетка місткістю 1,0 мл — 0,60%, мірна колба місткістю 50 мл — 0,17%.
ЛП (sample): наважка 10 мг ЛП Z — 0,2 мг або 100·0,2/10 = 2,00%, мірна колба місткістю 10 мл — 0,50%.
У цьому випадку при використанні лінійної моделі, беручи до уваги рівняння (1, 3), отримаємо граничну невизначеність пробопідготовки:
 (4)
(4)
Ця невизначеність, природно, не враховує суб’єктивного фактора — кваліфікації аналітика, який може бути в окремих випадках дуже значущим.
2.2. Прогноз граничної невизначеності кінцевої аналітичної операції (∆FAO). Рідинна хроматографія. Гранична невизначеність кінцевої аналітичної операції (∆FAO) визначається процедурою виконання методики (кількістю паралельних вимірів тощо) та вимогами до обладнання. Величина ∆FAO зазвичай визначається вимогами до придатності хроматографічної системи. У нашому випадку RSDmax, розраховане з 5 хроматограм розчину порівняння, має бути не більше 0,90%. Можна вважати, що таке саме граничне RSD буде і для випробуваного розчину, який за концентрацією речовини, що аналізують, Х є близьким до розчину порівняння. Беручи до уваги співвідношення (3), гранична невизначеність
усієї кінцевої аналітичної операції буде:
 (5)
(5)
Множник √2 враховує прогнозовану рівність величин ∆FAO(st) і ∆FAO(sample).
Спектрофотометрія: величина RSDmax не регламентується тестом на придатність аналітичної системи, а визначається класом спектрофотометра і може сягати RSDmax= SSp,r=0,50%. Враховуючи це, рівняння (5) набуває вигляду:
 (6)
(6)
Отже, невизначеність спектрофотометрії у цьому випадку практично не відрізняється від рідинної хроматографії, що є типовим для сучасних хроматографів. У разі багатокомпонентної спектрофотометрії в чисельнику співвідношення (6) додається ще один множник — коефіцієнт підсилення спектрофотометричної похибки.
2.3. Прогноз граничної загальної невизначеності методики аналізу (∆As). Гранична невизначеність усієї методики аналізу проводиться за рівнянням (2).
Рідинна хроматографія:
 (7)
(7)
Спектрофотометрія:
 (8)
(8)
2.4. Порівняння ∆As з критичним значенням і висновок щодо метрологічної обґрунтованості методики аналізу. При визначенні метрологічної обґрунтованості методик фармацевтичного аналізу найважливішим аспектом є вироблення й застосування метрологічно обґрунтованих критеріїв прийнятності отриманої невизначеності. З цією метою можуть застосовуватися різні підходи, з яких найбільш прийнятним є підхід, що ґрунтується на принципі незначущості. Невизначеність методики аналізу є незначущою на рівні 95% порівняно з допусками вмісту речовини, що аналізується, якщо виконуються співвідношення:
для субстанції ΔАs ≤ BH — 100%. (9)
для ЛП  . (10)
. (10)
де H — верхня межа вмісту за специфікацією, у %; L — нижня межа вмісту за специфікацією, у %.
У загальному випадку систематична похибка методики (δ) повинна бути статистично незначущою. В той же час, згідно з принципом незначущості для конкретної методики, систематична похибка може бути значущою статистично (якщо метод дуже точний), але незначущою порівняно з граничною невизначеністю даної методики аналізу, тобто повинно виконуватися співвідношення:
 (11)
(11)
Граничне значення ∆As (величина max ∆As) визначається співвідношеннями (9, 10). При їх виконанні методику фармацевтичного аналізу можна вважати метрологічно обґрунтованою. В іншому разі — ні. Зокрема, у нашому випадку аналізу ЛП Z: BH=105,0%, BL=95,0%. Тоді згідно з нерівністю (10) повинна виконуватися нерівність:
 (12)
(12)
Згідно з рівняннями (6, 7) гранична невизначеність аналізу і рідинної хроматографії й спектрофотометрії становить 2,5%, тобто нерівність (12) не задовольняється для обох методів, і обидві методики аналізу є метрологічно необґрунтованими. Головною причиною цього є дуже велика невизначеність пробопідготовки (2,20%), яка викликана незадовільною процедурою її виконання.
На відміну від невизначеності кінцевої аналітичної операції, яка значною мірою визначається рівнем обладнання, невизначеність пробопідготовки може бути мінімізована вибором наважок, мірних колб тощо. Зокрема, в нашому випадку слід замість наважки 10 мг (невизначеність 2,0%) ЛП Z у мірну колбу місткістю 10 мл (невизначеність 0,50%) брати наважку 50 мг (невизначеність 0,40%) у мірну колбу місткістю 50 мл (невизначеність 0,17%). У цьому випадку рівняння (4) для прогнозу граничної невизначеності пробопідготовки має вигляд:
 (13)
(13)
Рівняння (7) для прогнозу граничної невизначеності всієї методики аналізу тоді має вигляд:
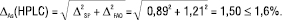
 (14)
(14)
У цьому випадку нерівність (12) виконується, і методику аналізу можна вважати метрологічно обґрунтованою. Беручи до уваги великий вплив невизначеності пробопідготовки на загальну невизначеність методики аналізу і можливість її мінімізування коригуванням процедури виконання, рекомендується планувати процедуру методик так, щоб невизначеність пробопідготовки була незначущою порівняно із загальною невизначеністю методики аналізу, тобто:
 (15)
(15)
У цьому разі невизначеність пробопідготовки можна не враховувати при прогнозі граничної невизначеності всієї методики аналізу.
3. Придатність хроматографічної системи. Термін «придатність системи» (System suitability test) був уведений Фармакопеєю США в 1990 р., має загальне значення і був рекомендований для всіх методів аналізу. Але інші методи аналізу досить добре стандартизовані і, як правило, не потребують додаткового введення цього тесту. Тому фактично він використовується лише для хроматографічних методів аналізу, які мають занадто багато нестандартизованих чинників, що впливають на його результати. Цей тест використовується для підтвердження, розділення й відтворюваності хроматографічної системи, є адекватним для вирішення аналітичної задачі. На сьогодні тест на придатність хроматографічної системи введений до всіх провідних світових фармакопей, включаючи ДФУ, та є обов’язковим для всіх хроматографічних методів.
Особливе значення тест має для кількісних хроматографічних методів аналізу, бо, як показано у рівнянні (5), граничне відносне стандартне відхилення площ (або висот) піків паралельних хроматограм (RSDmax) прямо впливає на невизначеність методики аналізу. Якщо хроматографічна система на момент аналізу неспроможна забезпечити необхідне значення RSDmax, отримані результати будуть мати неприйнятно велику невизначеність, тобто не будуть метрологічно обґрунтованими. Це може привести до різних висновків про якість ЛП, що аналізується.
Для вирішення питання, якою ж повинна бути ця прийнятна величина RSDmax, можуть застосовуватися різні підходи, що приведе до різних величин RSDmax. Статистично обґрунтованим можна вважати підхід, що базується на співвідношеннях (5–8). У загальному випадку невизначеність пробопідготовки є індивідуальною для кожної методики. Відповідно індивідуальними для кожної методики є й вимоги до RSD. У тому разі, коли виконується співвідношення (14) незначущості невизначеності пробопідготовки, задача значно спрощується, і величини RSD залежать тільки від допусків вмісту речовини, що аналізується, і числа паралельних хроматограм у тесті на придатність хроматографічної системи. Це дає можливість розрахувати максимальні прийнятні значення відносного стандартного відхилення для тесту на придатність хроматографічної системи (RSDmax).
Слід зазначити, що для субстанцій Європейська Фармакопея рекомендує вдвічі менші значення RSDmax. Можливо, це пов’язано з тим, що не висувається якихось вимог (подібних (14)) до невизначеності пробопідготовки.
4. Оптимізація кількості паралельних випробувань досить гостро стоїть у хроматографії, коли доводиться одночасно аналізувати велику кількість зразків, напр. при проведенні тестів «Розчинність», «Однорідність вмісту діючої речовини в одиниці дозованого ЛП» або при аналізі декількох серій ЛП. Враховуючи довготривалість однієї хроматограми (понад 20 хв), велику кількість зразків (до 30) і необхідність (таблиця) проведення значної кількості паралельних хроматограм, власне хроматографічний аналіз може тривати кілька діб. За такий час характеристики хроматографування можуть суттєво змінитися, що взагалі робить сумнівним отримання коректних метрологічних характеристик. Однак зменшення кількості паралельних хроматограм (n) призводить до збільшення невизначеності кінцевої аналітичної операції (∆FAO) і, відповідно, всієї методики аналізу в цілому (∆As) — за рахунок підвищення коефіцієнта t(95%, n–1) і зменшення знаменника √n у співвідношенні (6). Це збільшення особливо суттєве для найбільш практично важливих n (n=3 і менше) і може призвести до невиконання нерівності (9, 10), тобто методика аналізу й отримані результати стають метрологічно необґрунтованими.
Таблиця. Вимоги до RSDmax при проведенні кількісного визначення на етапі перевірки придатності хроматографічної системи (передбачається, що невизначеність пробопідготовки незначуща у порівнянні з повною невизначеністю методики аналізу)
| n | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| RSDmax, % | |||||||
| BH – 100% | Лікарська субстанція | ||||||
| 1 | 0,16 | 0,42 | 0,60 | 0,74 | 0,86 | 0,96 | 1,06 |
| 1,5 | 0,24 | 0,63 | 0,90 | 1,11 | 1,29 | 1,44 | 1,58 |
| 2 | 0,32 | 0,84 | 1,20 | 1,48 | 1,72 | 1,93 | 2,11 |
| 3 | 0,48 | 1,26 | 1,80 | 2,23 | 2,58 | 2,89 | 3,17 |
 |
ЛП | ||||||
| 5 | 0,25 | 0,67 | 0,96 | 1,19 | 1,38 | 1,54 | 1,69 |
| 7,5 | 0,38 | 1,01 | 1,44 | 1,78 | 2,06 | 2,31 | 2,53 |
| 10 | 0,51 | 1,34 | 1,92 | 2,37 | 2,75 | 3,08 | 3,38 |
| 15 | 0,76 | 2,01 | 2,88 | 3,56 | 4,13 | 4,62 | 5,07 |
| 20 | 1,01 | 2,68 | 3,85 | 4,75 | 5,50 | 6,16 | 6,76 |
Одним із шляхів вирішення цієї проблеми є використання об’єднаних відносних стандартних відхилень паралельних хроматограм усіх зразків, що аналізуються. Це пов’язано з близькістю в них концентрацій речовини, яку аналізують, що дає підстави вважати їх вибірками однієї й тієї ж генеральної сукупності. Раціональною схемою хроматографування N випробуваних зразків є: no хроматограм розчину порівняння (перевірка придатності хроматографічної системи; величина no визначається вимогами таблиці) — n хроматограм випробуваного розчину 1 — розчин порівняння –//– n хроматограм випробуваного розчину 2 — розчин порівняння; –//– n хроматограм випробуваного розчину і — розчин порівняння…, –//– n хроматограм випробуваного розчину N — розчин порівняння. Загалом отримують (N+no) хроматограм розчину порівняння і N·n хроматограм випробуваних розчинів 1…N. Розраховують відносні стандартні відхилення для всіх хроматограм розчину порівняння (RSDo) і для кожного з випробуваних розчинів (RSDi). Об’єднане відносне стандартне відхилення розраховується за співвідношенням:
 (16)
(16)
Невизначеність середнього результату аналізу (виражена як однобічний довірчий інтервал ∆As) для кожного випробуваного зразка визначають за рівнянням:
 (17)
(17)
Враховуючи співвідношення (9, 10), із рівнянь можна розрахувати граничні значення для величин RSD (значення RSDmax), на підставі яких і визначається мінімальне значення паралельних хроматограм n, яке дозволяє отримувати метрологічно обґрунтовані результати аналізу. Якщо no≤5, а кількість випробуваних зразків N>5, то для отримання метрологічно обґрунтованих результатів достатньо по 2 паралельних хроматограми для кожного випробуваного зразка. Це нерідко дозволяє в декілька разів зменшити об’єм хроматографічного експерименту і час аналізу без погіршення точності. Такий підхід запропонований для хроматографічних методів, але може бути застосований і для інших методів, напр. спектрофотометрії.
5. Отримання валідаційних характеристик методик фармацевтичного аналізу і їх метрологічна оцінка. Така задача є типовою при проведенні валідації методик кількісного визначення фармацевтичного аналізу. Характерною особливістю ЛП є досить вузькі допуски вмісту діючих речовин (зазвичай 5–10% номінального значення). Відповідно валідація методик кількісного визначення ЛП проводиться теж у досить вузьких межах (±20% від номінального значення). Крім того, більшість методик кількісного визначення (хроматографія, спектрофотометрія) є непрямими з використанням методу стандарту. Це дозволяє використовувати нормалізовані координати, в яких концентрацію й аналітичний сигнал виражено у відсотках до концентрації та аналітичного сигналу стандартного розчину, що дає можливість привести всі методики кількісного визначення до єдиного формату. При цьому доцільно об’єднати вивчення валідаційних характеристик лінійності, правильності й точності, що значно спрощує й стандартизує процедуру отримання та оцінки валідаційних характеристик методик аналізу. При такому підході відношення у відсотках (Х%) нормалізованого аналітичного сигналу до нормалізованої концентрації є відношенням у відсотках знайденої концентрації до введеної.
Обчислюючи величини Хі для всіх концентрацій, що вивчаються (раціональним є 9 концентрацій, рівномірно розподілених в інтервалі 80–120% номінальної концентрації), можна потім визначити середнє значення (X—), стандартне відхилення одиничного значення (sX), довірчий інтервал одиничного значення ΔX й довірчий інтервал середнього значення ΔX–. Величина ΔX=ΔAs є відносною випадковою невизначеністю аналітичної методики і повинна задовольняти співвідношення (9, 10). Величина │100 — X—│ = ő% є відносною систематичною похибкою методики і повинна бути або менше ΔX–, або (якщо це не виконується) задовольняти співвідношенню (11). Перевірка лінійності проводиться, як вказано в статті «Валідація аналітичних методик і випробувань».
Гризодуб А.И., Леонтьев Д.А., Доценко Т.Н., Денисенко Н.В. Метрологические аспекты официальных методик контроля качества лекарственных средств. 3. Выполнение теста «Количественное определение» при одновременном контроле качества нескольких образцов лекарственных средств хроматографическими методами // Фармаком. — 2002. — № 4; Гризодуб А.И., Леонтьев Д.А., Левин М.Г. Метрологические аспекты официальных методик контроля качества лекарственных средств. 1. Методики ВЭЖХ // Фізіологічно активні речовини. — 2001. — № 1 (31); ДФУ. 2.2.25. Абсорбційна спектрофотометрія в ультрафіолетовій і видимій областях. 2.2.28. Рідинна хроматографія. — Х., 2003; ДФУ. Доповнення 1. Статистичний аналіз результатів хімічного експерименту. Додатки до діючих текстів ДФУ 1. Валідація аналітичних методик і випробувань. D. Критерії проведення валідації для методик кількісного визначення. 2.2.25. Абсорбційна спектрофотометрія в ультрафіолетовій і видимій областях. 2.2.28. Рідинна хроматографія. — Х., 2004; European Pharmacopoeia. 4th edition. 4.8. 2.2.46. Chromatographic separation techniques. — 2003. — CD-ROM version.; The United States Pharmacopeial Convention. USP 22 — NF 17; The United States Pharmacopeial Convention. USP 24 — NF 19. <621> Chromatography.